

Keep up with the ever-evolving AI landscape
Unlock exclusive AI content by subscribing to our newsletter!!
ProtGPT2 generates sequences with properties ‘akin to their natural counterparts’
ProtGPT2 generates sequences with properties ‘akin to their natural counterparts’
Academics from the University of Bayreuth in Germany have thrown their hats into the growing ring for protein prediction AI models by unveiling ProtGPT2 — in a bid to accelerate drug discovery and better understand diseases.
The unsupervised language model is capable of generating protein sequences that follow similar principles found in naturally originating proteins, according to a paper published in Nature. These proteins generated by ProtGPT2 display natural amino acid propensities.
The authors’ findings show that 88% of ProtGPT2-generated proteins are globular — effectively, spherical proteins that have a wide range of functions inside a cell — in line with natural sequences. Rival models such as AlphaFold generate string-like proteins.
Protein predictions use language models because there are various similarities between the two. Protein sequences consist of a “chemically defined” alphabet, the authors explained. “These ‘letters’ arrange to form secondary structural elements (words), which assemble to form domains (sentences) that undertake a function (meaning).” Thus, advances in language models enable great strides in protein prediction models as well.
ProtGPT2, a transformer-based pre-trained model, boasts 738 million parameters and generates sequences that “show predicted stabilities and dynamic properties akin to their natural counterparts,” the authors wrote.
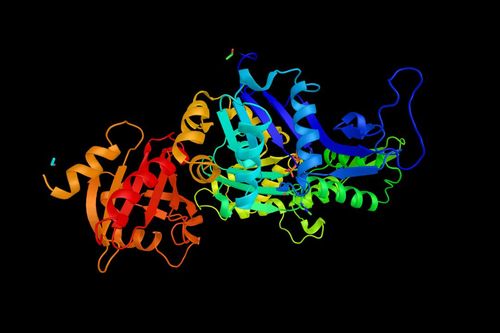
Figure 1:  Examples of proteins generated by ProtGPT2 (Image credit: Nature)
Examples of proteins generated by ProtGPT2 (Image credit: Nature)
The globular proteins predicted by ProtGPT2 act as enzymes, or stocks of amino acids, and messengers, by transmitting messages to regulate biological processes like hormones.
“Since protein design has an enormous potential to solve problems in fields ranging from biomedical to environmental sciences, we believe that ProtGPT2 is a timely advance towards efficient high-throughput protein engineering and design,” the paper reads.
The model and datasets are available via HuggingFace and has already been downloaded over 4,300 times.
Scientists believe that the ability to predict a protein’s 3D structure could enable faster drug discovery thanks to a better understanding of how the body’s proteins relate to diseases.
Each cell in the human body contains billions of proteins that control vital functions. Those proteins contain amino acids arranged in formations like strings or spheres. Those formations fold themselves into a 3D shape based on the interactions of these amino acids, which then perform different tasks in the body, such as carrying oxygen in the blood from the lungs to body tissues.
Here is an explainer from DeepMind about the concept:
There are several competing AI-powered protein prediction models around.
Arguably the most famous is AlphaFold, developed by Google-owned DeepMind. The deep-learning neural network has 21 million parameters and was trained on more than 170,000 proteins from a public repository of protein sequences and structures.
The system itself uses an attention network − a deep learning technique where an algorithm recognizes parts of a larger problem — then pieces them together to obtain the overall solution. It can do this in minutes or hours, depending on the size of the protein.
In late July, DeepMind published predicted structures for 200 million proteins, which it claimed represent “nearly all cataloged proteins known to science.”
Related stories:
DeepMind challenger? A ‘super fast' protein predicting model
DeepMind AI system predicts structure of nearly all known proteins
Meta challenges DeepMind with protein-folding AI model
DeepMind and EMBL release massive database of AI-based human protein structure predictions
DeepMind releases AlphaFold 2—an open source version of its protein-folding AI system
DeepMind achieves significant protein folding breakthrough using AI
In a challenge to DeepMind’s crown, AI researchers from Meta recently released ESMFold — a rival model that reportedly boasts “competitive” accuracy levels with AlphaFold.
ESMFold boasts 15 billion parameters and can accurately predict full atomic protein structures from a single sequence of a protein.
In a bid to rival AlphaFold, Meta is planning to open source ESMFold in the future.
Another challenger model recently emerged from Chinese biotech company HeliXon, which works in the drug discovery space.
Their model, OmegaFold, outperformed rival model RoseTTAFold while achieving similar prediction accuracy to AlphaFold2, according to a paper from the Chinese company.
OmegaFold can predict high-resolution protein structure from a single primary sequence. The researchers claim in their paper that they retrained an AlphaFold2 model inputting only single sequences and found that it was “not able to match the prediction quality of OmegaFold.”
You May Also Like
.jpg?width=700&auto=webp&quality=80&disable=upscale)
.jpg?width=700&auto=webp&quality=80&disable=upscale)
.jpg?width=700&auto=webp&quality=80&disable=upscale)
.jpg?width=300&auto=webp&quality=80&disable=upscale)
.jpg?width=300&auto=webp&quality=80&disable=upscale)
.jpg?width=300&auto=webp&quality=80&disable=upscale)

