

Keep up with the ever-evolving AI landscape
Unlock exclusive AI content by subscribing to our newsletter!!
AlphaFold2 rival trained on as many as 15 billion parameters.
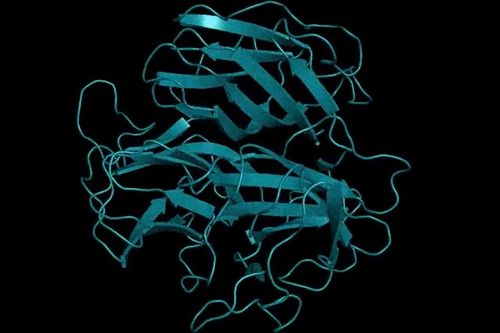
AlphaFold2 rival trained on as many as 15 billion parameters
Researchers from Meta created an AI model that can accurately predict full atomic protein structures from a single sequence of a protein – helping to solve one of biology’s biggest challenges and taking on DeepMind’s ground-breaking model AlphaFold2.
Each living cell contains billions of proteins, which are molecular machines that control a body’s vital cell functions. And each protein consists of a string of amino acids arrayed like a necklace. A protein string ‘folds’ itself into a 3D shape based on the interactions of these amino acids, which then perform different tasks in the body such as carry oxygen in your blood from the lungs to body tissues.
Predicting a protein’s 3D structure could help to speed up drug discovery by replacing “slow, expensive” structural biology experiments with faster, cheaper computer simulations, among other benefits, according to research by the National Institutes of Health. It also helps scientists better understand the body especially since misfolded or abnormal proteins can lead to diseases.
Meta’s protein folding model is called ESMFold, or Evolutionary Scale Modeling. It was unveiled by Meta research scientist Alex Rives in a Twitter thread where he suggested the model’s accuracy levels were “competitive” with AlphaFold2. DeepMind’s model had a median accuracy of 92.4 Global Distance Test, which gauges the difference between predicted and the reference structure. The highest score is 100.
“ESMFold has similar accuracy to AlphaFold2 and RoseTTAFold for sequences with low perplexity that are well understood by the language model,” a research paper covering the new model reads. “ESMFold inference is an order of magnitude faster than AlphaFold2, enabling exploration of the structural space of metagenomic proteins in practical timescales.”
Meta plans to open source ESMFold in the future, according to research engineer Zeming Lin via Twitter.
Notably, Meta’s researchers did publish several pretrained language models for proteins on GitHub under an MIT license.
The Meta team took a page from large language models – such as Gato, GPT-3 and Bloom – that once scaled were able to yield new capabilities such as higher level reasoning and generating lifelike images.
While protein sequences have been used for training of language models, they were done on a smaller scale. With ESMFold, Meta trained proteins on large language models up to 15 billion parameters to see what it yielded. The team believe these are the “largest language models of proteins to be evaluated to date.”
The results? The team discovered that ESMFold yielded “high accuracy end-to-end atomic level structure prediction directly from the individual sequence of a protein.”
According to Lin, the three billion-parameter language model took three weeks to develop on 256 GPUs, and ESMFold took 10 days on 128 GPUs. The 15-billion version took around 45 days to train using 800 GPUs – though the research team suggested that GPU utilization was worse due to a slow internode connection.
Meta’s team found that as models are scaled, they learn information enabling the prediction of the three-dimensional structure of a protein at the resolution of individual atoms.
“As ESM2 processes a protein sequence, a picture of the protein’s structure materializes in its internal states that enables atomic resolution predictions of the 3D structure, even though the language model was only trained on sequences,” Rives explained.
“There are billions of protein sequences with unknown structure and function, many from metagenomic sequencing. ESMFold makes it feasible to map this structural space in practical timescales. We were able to fold a random sample of one million metagenomic sequences in a few hours.”
According to Rives, who also co-founded biotech company Fate Therapeutics, the newly unveiled model can “help to understand regions of protein space that are distant from existing knowledge.”
The model Meta is challenging through ESMFold is Alphafold2, a deep-learning neural network that can accurately determine a protein’s 3D shape from its sequence of amino acids. The initial version was released in 2018, with Google-owned DeepMind publishing a second version in 2020 and a year later released the source code.
Also rivaling ESMFold is RoseTTaFold, an open source model developed by a biochemist team from the University of Washington.
AlphaFold2 can generate 3D structures of models slightly faster than RoseTTaFold, creating structures in minutes or hours, depending on the size of the protein. However, Meta’s AI team claims ESMFold’s inference speeds are faster than AlphaFold2. Meta also contends its model produces “more accurate atomic-level predictions” than AlphaFold2 or RoseTTAFold when they are artificially given a single sequence as an input.
You May Also Like
.jpg?width=700&auto=webp&quality=80&disable=upscale)
.jpg?width=700&auto=webp&quality=80&disable=upscale)
.jpg?width=700&auto=webp&quality=80&disable=upscale)
.jpg?width=300&auto=webp&quality=80&disable=upscale)
.jpg?width=300&auto=webp&quality=80&disable=upscale)

.jpg?width=300&auto=webp&quality=80&disable=upscale)
.jpg?width=300&auto=webp&quality=80&disable=upscale)